工作原理
不同于暴力枚举,CSD 直接在组合化学空间中智能对接
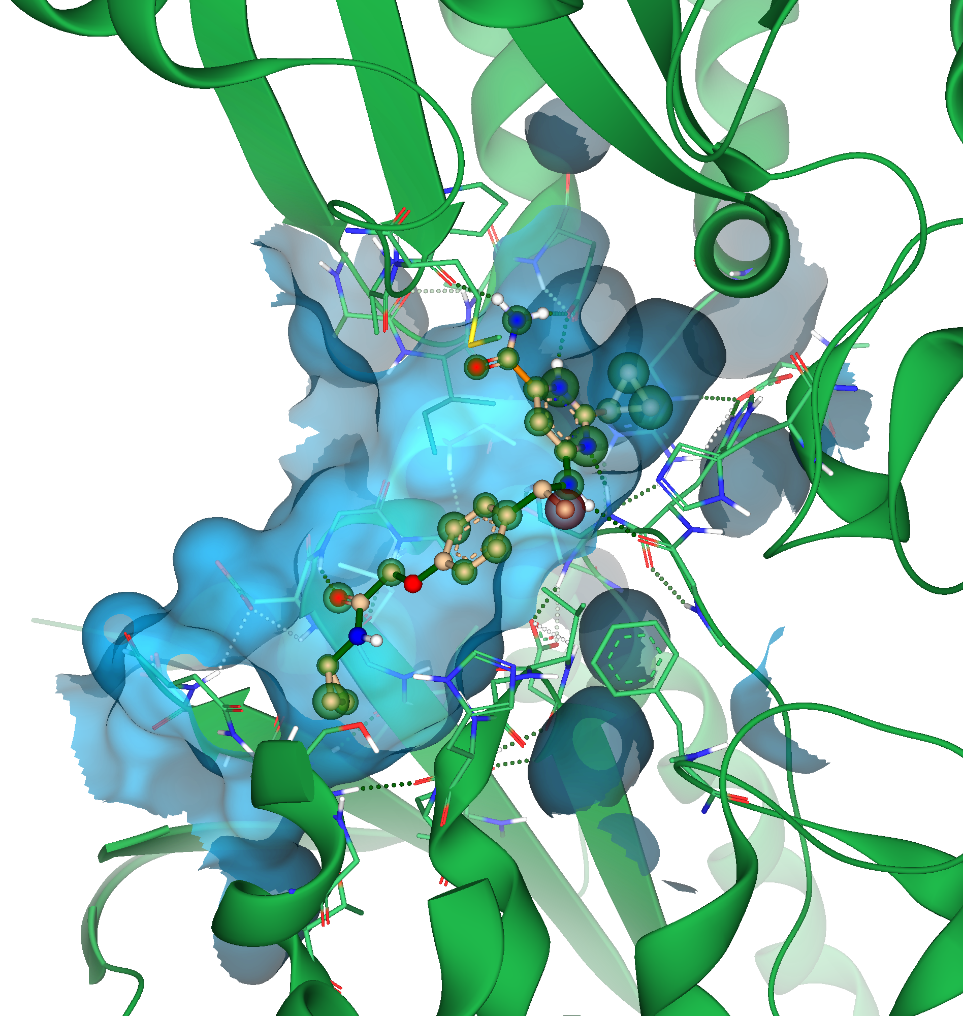
合成子锚定
将含有延伸向量的砌块(合成子)对接到靶标结合位点,筛选出与靶标形成高质量相互作用的最佳候选。
第一轮延伸
基于选中的合成子,枚举所有化学可及的化合物并进行对接。低分合成子已被丢弃,大幅节省算力。
第二轮延伸
部分化合物在第一轮后即完成;其余进行第二轮延伸生成最终分子。所有结果均为可合成化合物。
核心思路:只枚举有前途的分支,跳过无关组合。评估万亿分子所需算力仅为传统方法的极小部分。
核心优势
不只是虚拟筛选,是下一代增强型先导物发现
数据不出门
在您自己的硬件上运行,无需将数据共享给第三方云服务或外部服务商。完全本地部署,自主可控。
轻量硬件即可
无需昂贵的 GPU 集群或超级计算机。普通 CPU 服务器即可实现高计算效率,大幅降低基础设施成本。
化学多样性
方法天然检索互补不同子口袋的多样化学,为您的管线提供丰富的骨架多样性。
结果可合成高达 80%+
化学空间的组合构建方式保证所有命中分子均为可合成化合物,可直接向合作供应商下单采购。
易于使用
从 SeeSAR 的可视化界面中直接操作,每个步骤都清晰直观,专家和新手均可快速上手。
多化学空间支持
支持多个商业化学空间,每个空间有独立的砌块和反应体系,覆盖不同的化学领域。
已发表案例
来自顶级药企和研究机构的实证数据
Genentech — ROCK1 激酶抑制剂
与 Genentech 合作,筛选 ROCK1 激酶的抑制剂。69 个采购化合物中,27 个 (39%) Ki < 10 µM。两个先导物的共晶结构与对接预测高度一致。
Crystals First — PKA 片段优化
从 4 个小分子片段出发筛选 PKA 新型抑制剂。93 个合成化合物中,40 个 (43%) 有活性。最佳片段优化物亲和力提升 13,500 倍,获得 6 个晶体结构。全程仅需 9 周。
技术细节
精雕细琢的每一个环节
无偏对接
CSD 对接真实合成子,在砌块中引入无偏延伸向量("虚拟原子")进行结合模式预测,而非依赖最小产物。
模板支持
使用共晶结构或对接预测作为模板,加速和引导锚定步骤的位姿生成,特别适合 FBDD 场景。
碰撞采样
合成子的虚拟原子在延伸步骤中扫描靶标表面,预先排除会导致位阻冲突的位姿,减少无效计算。
可用化学空间
与全球主要化合物供应商合作,命中分子数周内交付
HPSee — 算力扩展
高性能计算工作流管理器
当本地硬件不够用时,通过 HPSee 将 CSD 任务无缝扩展到远程 HPC 集群。一键提交,完成后只下载排名靠前的结果在 SeeSAR 中评估。
- 化合物库管理 — 集中管理远程化合物集合
- 团队协作 — 多人共享计算资源和结果
- 工作流编排 — 标准化虚拟筛选流程